Механизм радикальной полимеризации на примере. Инициирование радикальной полимеризации
2.1.1. Инициирование радикальной полимеризации
Процесс инициирования является первым шагом свободно-радикальной полимеризации:
Первичные радикалы, необходимые для инициирования радикальной полимеризации, могут быть получены в результате химических реакций и при физическом воздействии на мономер.
Вещественное инициирование. При химическом или вещественном инициировании используют вещества, распадающиеся с образованием свободных радикалов, или смеси веществ, реагирующих между собой с образованием свободных радикалов. В качестве таких веществ обычно используют пероксиды и азосоединения, а также комбинации веществ, образующих окислительно-восстановительную систему.

Среди пероксидов широкое применение нашли ацил-, алкил-, гидропероксиды и перэфиры. Круг азосоединений, практически используемых в качестве инициаторов, более ограничен. Наиболее известным среди них является 2,2"-азобис(изобутиронитрил), распадающийся с выделением азота:

Благодаря последнему обстоятельству, этот и подобные ему азопроизводные используются в промышленности не только как инициаторы, но и для вспенивания пластмасс при получении пенопластов.
Наиболее употребляемые в современной исследовательской и производственной практике инициаторы приведены в табл. 2.1 наряду с характеристиками их распада. Таблицу замыкают высокотемпературные инициаторы, распадающиеся с разрывом связи С-С.
Окислительно-восстановительные системы делятся на две группы; органо- и водорастворимые. К первой группе относятся многочисленные комбинации пероксидов с аминами, из которых наиболее изученной является система пероксид бензоила – диметиланилин. В результате протекания окислительно-восстановительной реакции в этой системе, первичным актом которой является передача электрона от амина к пероксиду, образуется бензоат-радикал, который и инициирует далее процесс полимеризации:
Таблица 2.1
Важнейшие инициаторы радикальной полимеризации
|
Инициатор |
Е расп, кДж/моль |
А расп, с -1 |
Температура, ºС для τ 1/2 |
||
|
Бис(2-этилгексил)пероксидикарбонат
| |||||
|
| |||||
|
(изобутиронитрил)
| |||||
|
Пероксид бензоила | |||||
|
пероксибензоат | |||||
|
| |||||
|
пероксид
| |||||
|
Гидропероксид кумила | |||||
|
Дициклогексилпероксидикарбонат
| |||||
|
3,4-Диметил-3,4-дифенилгексан
| |||||
|
2,3-Диметил-2,3-дифенилбутан
| |||||

 Пероксид
лаурила
Пероксид
лаурила 2,2"-Азобис
2,2"-Азобис
 трет
-Бутил
трет
-Бутил Пероксид
кумила
Пероксид
кумила Ди-трет
-бутил
Ди-трет
-бутил




В рассматриваемом примере образование окислительно-восстановительной системы приводит к увеличению скорости полимеризации и снижению температуры ее инициирования по сравнению с процессом, инициируемым лишь одним пероксидом. Эти преимущества характерны и для других окислительно-восстановительных систем.
Водорастворимые окислительно-восстановительные системы берут начало от классической системы:
часто называемой реактивом Фентона. Они могут быть образованы и другими ионами металлов переменной валентности и пероксидами. Вместо последних в водных растворах обычно используют гидропероксиды.
Наибольшее распространение к настоящему времени получили окислительно-восстановительные системы, содержащие в качестве окислителя персульфаты, а в качестве восстановителя – ионы металла переменной валентности или тиосульфаты:
Они широко применяются в промышленности для инициирования эмульсионной и растворной полимеризации.
Для правильного выбора инициатора полимеризации необходимо располагать данными, характеризующими скорость его распада при температуре реакции. Наиболее универсальной характеристикой является период полураспада инициатора τ 1/2 (табл. 2.1.). Чаще всего инициатор характеризуется при температуре, при которой время полураспада составляет 10 часов. Эта температура находится обычно в интервале от 20 до 120ºС и зависит от структуры инициатора. Обычно для инициирования полимеризации используют инициаторы, период полураспада которых соизмерим с продолжительностью процесса. Поскольку для реакций первого порядка τ 1/2 = ln 2/ k расп , то, зная величину τ 1/2 , можно рассчитать концентрацию инициатора в любой момент полимеризации в соответствии с уравнением:
 , (2.1)
, (2.1)
где k расп - константа скорости мономолекулярной реакции распада инициатора;
[ I 0 ] и [ I ] - начальная и текущая концентрации инициатора.
Фотохимическое инициирование. При облучении мономера УФ-светом молекулы, поглотившие квант света, возбуждаются и затем распадаются на радикалы, способные инициировать полимеризацию:
M + hν → M * → R 1 · + R 2 ·
Однако прямое облучение мономера малоэффективно, поскольку кварцевое стекло обычно не пропускает УФ-свет в области, соответствующей его поглощению мономером (π-π* -переход, 200-220 нм), или пропускает его в незначительной степени.
В том случае, когда мономер не поглощает прошедший свет, необходимо использовать фотосенсибилизатор (Z) - соединение, передающее энергию возбуждения другим молекулам:
Z + hν → Z *
Z * + M → Z + M * → R 1 · + R 2 · + Z .
Применение в качестве фотосенсибилизаторов красителей позволяет использовать для фотоинициирования видимую область света.
В практических целях фотополимеризация обычно проводится в присутствии фотоинициаторов - веществ, распадающихся в требуемой области УФ-спектра с достаточно высоким квантовым выходом. Идеальный фотоинициатор должен отвечать следующим критериям:
1. во-первых, разлагаться при облучении источником света с определенной длиной волны, которая не поглощается мономером;
2. эффективность инициатора должна быть высокой и близкой к единице;
3. лучше, если при этом образуется один тип радикалов.
По механизму действия фотоинициаторы можно разделить на 2 типа: распадающиеся на радикалы при облучении, например, соединения, содержащие бензольную группу (ацетофенольный тип) (I ) и взаимодействующие с соинициаторами с образованием радикалов (азосоединения (II )).

Механизмы действия азосоединений имеют особенности, которые заключаются в изменении при облучении своей конфигурации с цис- на транс- . Это имеет особое значение при импульсном воздействии света.

Наиболее эффективными фотоинициаторами являются ароматические кетоны и их производные, благодаря достаточно широкой области поглощения УФ-спектра и высокому квантовому выходу радикалов. Считается, что ароматические кетоны претерпевают фотохимическое превращение по двум направлениям:
последнее из которых реализуется лишь в присутствии доноров водорода.
В промышленности в качестве фотоинициаторов используют бензоин (1), бензилкеталь (2) и их многочисленные производные:

Зная количество поглощенных фотонов (n abs ) и облучаемый объем (V ), можно определить концентрацию первичных радикалов (С ), образующихся при монохроматическом облучении светом:
 , (2.2)
, (2.2)
где Ф – первичный квантовый выход.
Количество же поглощенных фотонов можно вычислить по известному выражению Ламберта-Бера:
 ,
(2.3)
,
(2.3)
где E p – поглощенная энергия, E λ – энергия одного моля фотонов при длине волны λ , - коэффициент экстинкции, С – концентрация, d – оптическая плотность.
Фотополимеризация используется для нанесения полимерных покрытий непрерывным способом на металл, дерево, керамику, световоды, в стоматологии для отверждения композиций зубных пломб. Особенно следует отметить применение фотополимеризации в фотолитографии, с помощью которой изготавливают большие интегральные схемы в микроэлектронике, а также печатные платы (матрицы) в современной технологии фотонабора, позволяющей исключить использование свинца.
Основное преимущество фотоинициирования в процессах полимеризации – возможность точного определения начала и завершения процесса через продолжительность воздействия света. Кроме того скорость разложения инициатора практически не зависит от температуры, в то время как интенсивность облучения вносит решающий вклад.
Существенным недостатком фотоинициирования является быстрое падение его эффективности с увеличением толщины облучаемого слоя вследствие поглощения излучения. По этой причине фотохимическое инициирование эффективно при возбуждении полимеризации в достаточно тонких слоях, порядка нескольких миллиметров.
Радиохимическое инициирование. Излучение радиоактивных источников Со 60 , а также различного рода ускорителей включает набор частиц, таких как -частицы, нейтроны, электроны и жесткое электромагнитное излучение. В отличие от фотоизлучения радиоактивное является ионизирующим и обладает гораздо большей проникающей способностью, что объясняется большей энергией его частиц.
Ионизация облучаемого вещества является следствием выбивания электронов из его молекул, например мономера, частицами высокой энергии:
Радикалы, способные инициировать полимеризацию, возникают в результате дальнейших превращений в системе с участием возбужденных ионов, ионрадикалов и электронов, например:

Наличие в облученном мономере свободных радикалов и ионов предопределяет возможность развития как радикальной, так и ионной полимеризации. В большинстве случаев результатом является радикальная полимеризация, однако, при низкой температуре в отсутствие воды и других примесей, дезактивирующих ионы, удалось наблюдать как катионную, так и анионную полимеризацию отдельных мономеров.
Термическое инициирование. Имеется очень мало примеров термического инициирования полимеризации. К ним относятся, прежде всего, спонтанная полимеризация стирола и винилпиридинов. Считается, что механизм возникновения свободных радикалов при термическом инициировании является бимолекулярным, но достаточно надежно он выявлен лишь по отношению к стиролу. Первой стадией реакции является образование аддукта Дильса-Альдера из двух молекул стирола:

На второй стадии имеет место перенос атома водорода от аддукта к молекуле стирола, что и приводит к возникновению радикалов, способных инициировать полимеризацию:
Самоинициированная полимеризация стирола имеет большую энергию активации. Так 50% конверсия мономера наблюдается при 29ºС в течении 400 дней, при 127ºС реакция проходит в течение 4 часов. Достоинство такого способа в том, что конечные полимеры не содержат примесей инициатора.
В большинстве других случаев спонтанная термическая полимеризация обусловлена инициированием перекисями, которые легко образуются на свету даже при кратковременном контакте мономеров с кислородом воздуха.
Инициирование плазмой. Здесь также как и в предыдущем случае образуются ионы и радикалы. Процесс полимеризации при этом протекает сложно. Данный метод используется при получении тонких полимерных пленок.
Электроинициирование. Протекает при электролизе смеси, содержащей органический растворитель, мономер и неорганическое соединение, проводящее электрический ток. При этом образуются ионы и радикалы.
Эффективность инициирования. Эффективность инициирования f равна доле радикалов, инициирующих полимеризацию, от их общего числа, которое соответствует спонтанному распаду определенного количества инициатора. Обычно 0,3 < f < 0,8, т.е. заметно меньше единицы. Это объясняется двумя причинами – индуцированным распадом инициатора и побочными реакциями в «клетке».
Индуцированный распад инициатора происходит в результате его реакции с радикалом роста, т.е. в результате передачи цепи на инициатор. Это приводит к уменьшению числа радикалов распавшегося пероксида, инициирующих полимеризацию:

Эффект «клетки» заключается в том, что два радикала, образовавшиеся в результате распада инициатора, в рассматриваемом случае пероксида бензоила, не могут в течение некоторого времени разойтись, поскольку их диффузии препятствуют окружающие молекулы мономера и растворителя. Этот момент весьма благоприятен для протекания побочных реакций, приводящих к их дезактивации. Одна из них приведена ниже (радикалы в «клетке» обозначены скобками):
Первичные бензоатные радикалы покидают «клетку» путем диффузии и в результате реакции с мономером. Далее они могут декарбоксилироваться
в результате чего реакция с мономером (инициирование) осуществляется с участием как бензоатных, так и фенильных радикалов:

К побочным реакциям, снижающим эффективность инициирования, помимо приведенной выше реакции в «клетке», относятся следующие две реакции:

В общем случае эффективность инициирования определяется природой инициатора, мономера, растворителя и конверсией. Большое значение имеет микровязкость среды, т.е. вязкость мономера или смеси мономер растворитель. Она определяет подвижность «клетки»: с ее увеличением выход радикалов из «клетки» затрудняется, и эффективность инициирования падает. Еще в большей степени уменьшается эффективность инициирования с увеличением конверсии, т.е. доли мономера, превратившегося в полимер.
Радикальная полимеризация, как правило, представляет собой разновидность цепных реакций. Такие реакции протекают под влиянием свободных радикалов, образующихся в начале процесса и реагирующих далее с нейтральными молекулами с образованием новых реакционноспособных радикалов.
Цепная полимеризация может инициироваться методами, известными для газофазных цепных реакций, в том числе ультрафиолетовым излучением. Один акт инициирования цепной полимеризации ведет к соединению друг с другом тысяч мономерных молекул. Другими признаками радикального цепного характера реакции полимеризации являются влияние примесей и формы реакционного сосуда на ее скорость, специфический s-образный вид кинетической кривой (зависимость степени превращения мономера в полимер от времени, рис. 5).
|
|
Рис. 5. Типичная кинетическая s-образная кривая полимеризации
Радикальная полимеризация имеет три характерные для цепных реакций стадии: инициирование, рост и обрыв цепи.
Для инициирования
реакции необходимо, чтобы в системе осуществилось получение (генерирование) свободных радикалов в результате теплового воздействия (термическое инициирование),
светового (фотоинициирование),
радиоактивного облучения (радиационное инициирование),
введение химических инициаторов (химическое радикальное инициирование)
и др. Термическое инициирование применяется редко, так как связано с большими затратами энергии, и при этом плохо поддаются регулированию как сам процесс реакции, так и свойства готового полимера. Фотоинициирование применяется главным образом для изучения механизма реакций полимеризации. Оно состоит в возбуждении молекулы мономера в результате поглощения кванта света и в генерировании затем свободных радикалов. В отличие от термической полимеризации скорость фотополимеризации не зависит от температуры, так как энергия активации ее значительно ниже. Скорость растет с увеличением интенсивности облучения. В этом случае подтверждением цепного характера реакции является протекание полимеризации после удаления источника света (рис. 6).

Рис. 6. Скорость полимеризации бутадиена: 1 - при освещении, 2 – после прекращения освещения
Радиационная полимеризация в принципе аналогична фотополимеризации. Скорость ее также растет с увеличением интенсивности облучения и не зависит от температуры. Скорость радиационной и фотополимеризации может быть увеличена добавлением веществ, которые легко распадаются под действием радиационного излучения или света (так называемые сенсибилизаторы полимеризации), например полигалогениды - CCl4, C2Cl6 и др.
Термический, фото - и радиационный способы инициирования цепной реакции полимеризации либо мало эффективны, либо сопровождаются протеканием различных побочных явлений (разветвление, деструкция цепей и т. д.). Поэтому на практике чаще всего применяется химическое инициирование, которое осуществляется специально вводимыми в систему легко распадающимися на радикалы веществами - инициаторами. Наиболее распространены среди них перекиси, азо - и диазосоединения. Распад этих соединений на радикалы может быть осуществлен различными путями, включая нагревание, фотохимическое разложение и др. Например, при легком нагревании перекись бензоила распадается по схеме
а гидроперекись изопропиленбензола так:
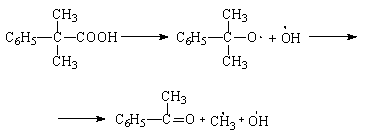
Динитрил азоизомасляной кислоты (азо-бис-изобутиронитрил) распадается с выделением азота:
Свободные радикалы (R·) легко реагируют с молекулой мономера:
|
|
которая становится свободным радикалом и реагирует со следующей молекулой мономера, и таким образом осуществляется реакция роста цепи. Поскольку стабильность радикалов, образующихся при распаде перекисей, азосоединений и других инициаторов, разная, то и скорость их реакции с молекулами мономера, а следовательно, и скорость полимеризации различны. Для облегчения распада инициаторов и снижения энергии активации стадии инициирования в реакцию вводят восстановители (амины и другие соединения, соли металлов переменной валентности).
Стадия роста цепи требует значительно меньшей энергии активации - 25,1-33,5 кДж/моль (6-8 ккал/моль), чем стадия инициирования - 84-126 кДж/моль (20-30 ккал/моль), и представляет взаимодействие растущих свободных радикалов с молекулами мономера, что приводит в итоге к образованию макромолекулы полимера:
Нейтральная макромолекула образуется на стадии обрыва цепи, энергия активации которой 8-17 кДж/моль (2-4 ккал/моль):

Такой обрыв цепи происходит в результате столкновения двух растущих макрорадикалов (рекомбинация). Возможно также диспропорционирование таких радикалов с образованием двух нейтральных молекул:
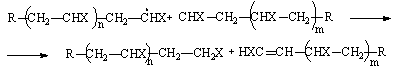
Причиной обрыва цепи может быть также присоединение к макрорадикалу низкомолекулярных веществ, присутствующих в системе (инициаторы, ингибиторы и др.). Время жизни растущих радикалов мало (обычно несколько секунд). По мере роста радикалов увеличивается вязкость системы, и вследствие уменьшения подвижности макрорадикалов скорость обрыва цепи путем рекомбинации снижается. Время жизни радикалов возрастает также при снижении температуры. Рост времени жизни макрорадикалов при увеличении вязкости системы приводит к интересному явлению - ускорению полимеризации на поздних стадиях (гель-эффект) вследствие увеличения концентрации макрорадикалов.
Как можно видеть из приведенных схем реакций роста и обрыва цепи, образуются макромолекулы полимера разной молекулярной массы. Широкий разброс значений молекулярной массы для образца полимера обычно приводит к ухудшению его механических свойств. Поэтому при получении полимера стремятся регулировать его молекулярную массу, что можно осуществить путем направленного изменения скорости роста цепи.
Для этой цели пользуются реакцией передачи цепи, которая заключается в том, что вводимое в систему вещество - регулятор - обрывает растущую цепь, но при этом само становится свободным радикалом и начинает новую кинетическую цепь реакции полимеризации. Таким образом, в данном случае обрывается материальная цепь, а кинетическая продолжается, в то время как в обычной реакции обрыва происходит обрыв как кинетической, так и материальной цепи. Роль агентов передачи цепи могут выполнять растворитель (особенно активны галогенсодержащие соединения, например СС14), мономер или специально вводимые вещества (регуляторы), например меркаптаны.
(обрыв цепи)
(начало новой цепи)
 или
или
Во всех случаях происходит рост новой макромолекулы полимера на каждый акт передачи цепи. Передача цепи может произойти также на молекулу полимера. В этом случае образуется разветвленная макромолекула. Повышение температуры и увеличение количества агента передачи цепи (например, галогенсодержащих углеводородов) приводят к резкому возрастанию скорости реакции передачи цепи, и эта реакция подавляет другие стадии полимеризации, так что образуются индивидуальные низкомолекулярные вещества, которые можно разделить (реакция теломеризации). Они содержат концевые группы из продуктов расщепления агента передачи цепи и являются активными в различных химических реакциях, в частности при получении новых полимеров.
Низкомолекулярные вещества, которые в результате реакции с радикалами мономеров предотвращают рост макромолекул или замедляют его, называются ингибиторами
или замедлителями.
Они широко используются для предотвращения преждевременной полимеризации или снижения ее скорости, для получения полимеров желательной молекулярной массы и более регулярной структуры. Такими веществами являются бензохинон, нитробензол и др. (рис. 7).
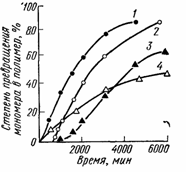
Рис. 7. Термическая полимеризация стирола при 100°С в присутствии ингибиторов и замедлителей:
1 - без добавок; 2- 0,1% бензохинона (ингибитор); 3 - 0,2% нитробензола (ингибитор); 4- 0,5% нитробензола (замедлитель)
Замедлитель выполняет двоякую роль: уменьшает концентрацию радикалов и время их жизни, что приводит к снижению длины полимерной цепи. Ингибитор не влияет на скорость полимеризации, но предотвращает начало инициирования цепи, увеличивая индукционный период на кинетической кривой полимеризации. Величина индукционного периода обычно пропорциональна количеству введенного ингибитора. Одно и то же вещество может выступать и как ингибитор, и как замедлитель, и как регулятор полимеризации - в зависимости от природы полимеризуемого мономера. В этом отношении особенно интересен кислород, который, например, замедляет полимеризацию винилацетата и ускоряет полимеризацию стирола. При больших давлениях и высоких температурах кислород способствует полимеризации этилена, что используется в промышленном производстве полиэтилена высокого давления. Кислород образует перекиси или гидроперекиси при взаимодействии с мономерами или растущими цепями. В зависимости от стабильности эти промежуточные перекиси или гидроперекиси могут либо увеличивать концентрацию радикалов и ускорять полимеризацию, либо дезактивировать имеющиеся радикалы и замедлять или даже ингибировать полимеризацию.
Рассмотрение кинетических закономерностей радикальной полимеризации дало возможность сделать ряд важных в практическом и теоретическом отношении выводов о влиянии различных факторов на этот процесс. Установлено, что скорость инициирования пропорциональна концентрации инициатора, а общая скорость полимеризации в стационарном периоде (когда скорость инициирования равна скорости обрыва цепи и, следовательно, общая скорость равна скорости роста цепи) пропорциональна квадратному корню из концентрации инициатора и первой степени концентрации мoномера u = K[M]1/2. Что касается степени полимеризации, т. е. молекулярной массы, то она обратно пропорциональна квадратному корню из концентрации инициатора n = K`[M]/1/2. Физический смысл этого положения заключается в том, что с ростом концентрации инициатора растет и число радикалов, образующихся в системе. Эти радикалы реагируют с большим числом молекул мономера и тем увеличивают скорость их превращения в растущие макрорадикалы. Однако при общем увеличении концентрации радикалов повышается и вероятность их столкновения друг с другом, т. е. обрыва цепи полимеризации. Это приводит к снижению средней молекулярной массы полимера.
Аналогичным образом можно рассмотреть влияние температуры на кинетику радикальной полимеризации. Обычно скорость полимеризации возрастает в 2-3 раза при повышении температуры на 10°. Повышение температуры увеличивает скорость инициирования полимеризации, так как облегчает распад на радикалы инициаторов и их реакцию с молекулами мономера. Вследствие большей подвижности малых радикалов с повышением температуры увеличивается вероятность их столкновения друг с другом (обрыв цепи путем диспропорционирования или рекомбинации) или с низкомолекулярными примесями (ингибиторами). Во всех случаях молекулярная масса полимера снижается, т. е. средняя степень полимеризации уменьшается с ростом температуры. Таким образом повышается количество низкомолекулярных фракций полимера в общем балансе распределения макромолекул по их молекулярным массам, возрастает доля побочных реакций, приводящих к образованию разветвленных молекул, появляется химическая нерегулярность построения цепи полимера вследствие увеличения доли типов соединения мономера «голова к голове» и «хвост к хвосту».
В радикальной полимеризации функции активных промежуточных продуктов (активных центров) выполняют свободные радикалы. В радикальную полимеризацию вступают мономеры с кратной С=С-связыо и мономеры с поляризованной кратной связью С=гетероатом. Циклические мономеры радикальной полимеризации не подвергаются.
К числу распространенных мономеров, вступающих в радикальную полимеризацию, относятся этилен, винилхлорид, винилацетат, винилиден- хлорид, тетрафторэтилен, акрилонитрил, метакрилонитрил, метилакрилат, метилметакрилат, стирол, бутадиен, хлоропрен и др. Перечисленные мономеры образуют высокомолекулярные продукты, в то время как виниловые эфиры и аллиловые мономеры образуют олигомеры. Виниленовые мономеры (СНХ=СПХ, за исключением X=F) в радикальную полимеризацию не вступают вследствие стерических затруднений.
Инициирование полимеризации - это превращение небольшой доли молекул мономера в активные центры (радикалы) под действием специально вводимых веществ (инициаторов) или излучения высоких энергий (радиационная полимеризация), или света (фотополимеризация) и др.
Наиболее распространенными методами инициирования являются термический гомолитический распад инициаторов, инициирование окислительно-восстановительными системами, фотохимическое инициирование, радиационное инициирование.
Термический гомолитический распад инициаторов осуществляется посредством инициаторов, к которым относятся различные типы перекисей: алкил- перекиси (перекись трет- бутила), гидроперекиси (гидроперекись кумола), перэфиры (от/д?ти-6утилпербензоат), ацилперекиси (перекись бензоила)
и азосоединения, среди которых наибольшее распространение получил 2,2"-азо-бмс-изобутиронитрил (ДАК или АИБН)

Эти инициаторы обычно нс отличаются селективным действием но отношению к разным мономерам, поэтому выбор инициатора чаще всего обусловливается температурой, при которой в каждом конкретном случае может быть достигнута желаемая скорость генерирования свободных радикалов. Так, ДАК применяют при 50-70°С, перекись бензоила - при 80-95°С, а перекись трет- бутила - при 120-140°С. Энергия активации инициирования обычно близка к энергии связи, разрывающейся при распаде инициаторов, и колеблется в пределах 105-175 кДж/моль.
Полимеризацию при высоких температурах можно вызвать и без введения в систему специальных инициаторов. В этом случае образование радикалов происходит, как правило, вследствие разложения небольших количеств пе- рекисных примесей, которые часто образуются при взаимодействии мономера с кислородом воздуха, или других случайных примесей. Возможность термического самоинициирования доказана только для ограниченного круга мономеров (стирола и некоторых его производных, метилметакрилата и ряда других).
Инициирование окислительно-восстановительными системами имеет преимущество - возможность осуществления полимеризации в водной или органической среде при комнатной температуре.
Приведем типичные окислительно-восстановительные инициирующие системы:
(также вместо солей железа используют соли Cr 2+ , V 2+ , Ti 3+ , Со 2 ")

Недостатком окислительно-восстановительного инициирования является низкая эффективность инициирования.
Фотохимическое инициирование протекает под действием УФ-света. В этом случае радикал может возникнуть как в системе, содержащей чистый мономер, так и при фотолитической диссоциации инициатора или в системе, содержащей фотосенсибилизатор, например бензофенон. Скорость фотоинициирования пропорциональна количеству поглощенного света. Удобство этого способа инициирования заключается в том, что процесс полимеризации можно вести при комнатной температуре.
Радиационно-химическое инициирование (под действием излучения высокой энергии) вызывает радикальную полимеризацию при температуре выше 0°С, а при пониженных температурах чаще происходит ионная полимеризация. К достоинствам этого процесса можно отнести легкость регулирования мощности дозы и времени полимеризации и высокую чистоту образующегося полимера.
Энергия активации фотохимического и радиационно-химического инициирования близка к пулю. Особенностью двух последних способов ииициирования является возможность мгновенного включения и выключения облучающего излучения.
Инициирование включает два элементарных акта:
а) генерирование радикалов R" из ининиатопа I:
![]()
б) взаимодействие радикала R* с мономером М:
![]()
Здесь & и и k" H - кинетические константы реакций инициирования.
Из этих двух стадий в большинстве случаев лимитирующей является стадия гемолитического распада инициатора, т.е. реакция (а).
Часть радикалов R" может расходоваться на побочные реакции, для учета этого вводят параметр «эффективность инициирования»/, равный отношению числа радикалов, участвовавших в реакции (б), к числу радикалов, образовавшихся по реакции (а).
Рост цени осуществляется последовательным присоединением молекул мономера к радикалам, возникающим в результате инициирования, например:
где k p - константа скорости роста цепи.
Развитие кинетической цепи сопровождается образованием материальной цепи макрорадикала. Значение энергии активации реакций роста цепи лежит в пределах 10-40 кДж/моль.
Константы скорости и энергия активации Е а реакции роста цепи в первую очередь зависят от природы мономера. Растворители, не склонные к специфическим взаимодействиям с молекулами мономера и растущими радикалами, не влияют на реакцию роста цепи радикальной полимеризации.
Энергия активации присоединения мономера к радикалу гем ниже, т.е. мономер тем активнее, чем выше энергия сопряжения в радикале, который получается в результате присоединения этого мономера к исходному радикалу. Таким образом, реакционные способности в ряду мономеров и соответствующих им радикалов изменяются антибатно.
Реакционная способность виниловых мономеров с заместителями уменьшается в ряду:
где R - алкил.
Реакционная способность соответствующих радикалов уменьшается справа налево.
К активным мономерам относятся мономеры, у которых двойная связь сопряжена с ненасыщенной группой заместителя, т.е. с большой энергией сопряжения. У неактивных мономеров сопряжение отсутствует или его (сопряжения) энергия мала. Чем выше реакционная способность мономера, тем выше энергия активации реакции роста цепи, т.е. тем ниже скорость его радикальной полимеризации.
Обрыв цепи приводит к ограничению кинетической и материальной цепи, т.е. к гибели активного центра (исчезновению активного радикала или его замене малоактивным радикалом, неспособным присоединять молекулы мономера). Обрыв цепи при радикальной полимеризации в основном происходит при взаимодействии двух растущих радикалов в результате их рекомбинации-.

где А и к ОЛ - кинетические константы обрыва по механизму рекомбинации и диспропорционирования соответственно.
Реакция обрыва цепи протекает в три стадии:
- 1) поступательная диффузия макрорадикалов с образованием объединенного клубка;
- 2) взаимное сближение активных концевых звеньев за счет сегментальной диффузии внутри объединенного клубка;
- 3) непосредственное химическое взаимодействие реакционных центров с образованием «мертвых» макромолекул.
Энергия активации реакции обрыва не превышает 6 кДж/моль и в основном определяется энергией активации взаимной диффузии радикалов.
В реакции обрыва цепи участвуют макрорадикалы разной длины, поэтому при полимеризации образуются макромолекулы разной длины (степени полимеризации). Конечный продукт полимеризации представляет собой полимер с широким молекулярно-массовым распределением.
Другой вариант обрыва цепи - обрыв на молекулах ингибитора. Ингибиторами могут быть малоактивные стабильные свободные радикалы (например, дифенилиикрилгидразил, N-оксидные радикалы), которые сами не инициируют полимеризацию, но способны рекомбинировать или диспро- порционировать с растущими радикалами. Ингибиторами также могут быть вещества, молекулы которых при взаимодействии с активными радикалами сами превращаются в малоактивные радикалы: это хиноны (бензо- хинон, дурохинон), ароматические ди- и тринитросоединения (например, динитробензол, тринитробензол), молекулярный кислород, сера и др. Ингибиторами служат также соединения металлов переменной валентности
(соли трехвалентного железа, двухвалентной меди и др.), которые обрывают растущие цепи за счет окислительно-восстановительных реакций. Часто ингибитор вводят в мономер для предотвращения его преждевременной полимеризации. Поэтому перед полимеризацией каждый мономер необходимо тщательно очищать от примесей и добавленного ингибитора.
В крайне редких случаях обрыв цепи может протекать мономолекуляр- но на стенках сосуда.
Передача цени также приводит к ограничению материальных цепей при полимеризации, но при этом активный центр не погибает, а переходит на другую молекулу. Реакции передачи цепи весьма характерны для радикальной полимеризации. Сущность этих реакций состоит в отрыве растущим радикалом атома или группы атомов от какой-либо молекулы (агента передачи цепи).
В качестве агента передачи цени может выступать специально добавленное к реакционной системе соединение с подвижным атомом или группой атомов, а также мономер, полимер или растворитель:

Здесь я м - кинетическая константа реакции передачи цепи на мономер; k u -кинетическая константа реакции передачи цепи на полимер; k s - кинетическая константа реакции передачи цепи на растворитель.
Отдельно следует отметить особенности полимеризации амиловых мономеров. В этом случае реакция передачи цепи на мономер с отрывом подвижного атома II в положении к двойной связи приводит к образованию резонансно-стабилизированного, неактивного аллильного радикала, не способного инициировать дальнейшую полимеризацию:
Аллильные радикалы рекомбинируют с образованием димеров. В этом случае, в отличие от обычной передачи, обрываются не только материальные, но и кинетические цени. Такой вид передачи получил название дегра- дационной передачи цепи. Деградационная передача, конкурируя с реакцией роста, приводит к крайне низким скоростям полимеризации аллиловых мономеров и образованию продуктов с невысокими молекулярными массами - олигомеров.
Склонность молекул мономеров участвовать в реакции передачи цепи принято характеризовать константой самопередачи
С м, равной отношению константы скорости реакции передачи цепи на мономер k M
к константе скорости реакции роста цепи k p:

Для большинства мономеров винилового ряда, не содержащих подвиж- н ых групп ил и атомов, k M k . Значение С ы обычно лежит в пределах 10" 4 -10 для аллиловых мономеров С м > 100 (табл. 5.6).
Таблица 5.6
Константы передачи при радикальной полимеризации
Способность растворителей участвовать в передачи цепи при радикальной полимеризации конкретного мономера характеризуют константой передачи-.

Реакции передачи цепи широко используются при синтезе полимеров для регулирования их молекулярных масс. Для уменьшения молекулярной массы синтезируемого полимера обычно применяют передатчики со значениями C s > 10 3 , которые называют регуляторами.
Кинетика радикальной полимеризации. Скорость инициирования при использовании термически распадающихся инициаторов можно выразить уравнением
где / - эффективность инициатора, которая обычно составляет от 0,5 до 1,0; ^р аспада - константа скорости распада инициатора; |1| - концентрация инициатора.
Скорость роста цепи V n выражается уравнением
где k-- константа скорости присоединения мономера к радикалу степени полимеризации г; | R* | - концентрация радикалов степени полимеризации г; [М] - концентрация молекул мономера.
Однако при образовании макромолекул большой молекулярной массы (степень полимеризации больше 5-10) можно считать, что k if) не зависит от степени полимеризации радикала. Тогда выражение для V p упрощается:
где | R* | - концентрация всех растущих радикалов.
С учетом допущения о том, что реакционная способность радикалов роста не зависит от степени их полимеризации, скорость исчезновения радикалов в результате реакции обрыва описывается уравнением
где k n - константа скорости обрыва.
Общая скорость полимеризации, равная скорости исчезновения мономера в системе, при условии, что степень полимеризации образующихся макромолекул достаточно велика и мономер расходуется только на полимеризацию, идентична скорости роста цепей, т.е.
Если в системе отсутствует ингибитор, то активные радикалы исчезают в результате их рекомбинации или диспропорционирования. В этом случае изменение концентрации радикалов описывается уравнением
Концентрацию радикалов , которую трудно измерить прямыми опытами, можно исключить из уравнения (5.10), приняв, что скорость образования радикалов равна скорости их исчезновения (условие квазистационарности), т.е. dR"]/dt = 0. При радикальной полимеризации это условие обычно выполняется уже через несколько секунд после начала реакции. Поэтому


В итоге получаем уравнение
Таким образом, допущения, необходимые и достаточные для вывода уравнения (5.11) скорости радикальной полимеризации, можно сформулировать следующим образом:
- 1) степень полимеризации должна быть много больше единицы;
- 2) константы элементарных стадий не зависят от степени полимеризации радикалов роста (принцип Флори);
- 3) если время жизни активных частиц мало по сравнению со временем полимеризации, то используют принцип квазистациопарпости, согласно которому изменение концентрации макрорадикалов во времени равно нулю, т.е. скорость инициирования равна скорости обрыва цепи;
- 4) процесс рассматривают на начальных конверсиях мономера.
Таким образом, порядок скорости реакции по концентрации мономера
составляет единицу, по концентрации инициатора - 0,5. Для того чтобы оценить влияние температуры на скорость полимеризации, рассмотрим суммарную энергию активации этого процесса. Эффективная константа скорости полимеризации
Тогда эффективная (суммарная) энергия активации процесса

Энергия активации реакции роста Е = HR40 кДж/моль, энергия активации реакции обрыва? 0 = (R6 кДж/моль, энергия активации реакции инициирования Е ИН = 105-Н75 кДж/моль для термического распада инициатора и Е нн = 0 для фото- или радиационного инициирования. Таким образом, в любом случае суммарная энергия активации реакции радикальной полимеризации положительна, и с ростом температуры скорость процесса возрастает.
Степень полимеризации. Из кинетических данных можно рассчитать длину кинетической цепи (v) и среднюю степень полимеризации (Р п) полученного полимера. Определим эти понятия.
Кинетическая цепь - число молекул мономера, приходящихся на один образовавшийся радикал R* до его гибели при обрыве цепи.
Таким образом, выражение для кинетической цепи имеет вид
При условии квазистационарности, используя уравнение (5.11), можно получить выражение
Материальная цепь (среднечисловая степень полимеризации) - число элементарных актов присоединения мономеров на один акт гибели радикала R’ при обрыве и передаче цепи.
При обрыве диспропорционированием (& од) одна макромолекула образуется из одной кинетической цепи, и длина материальной цепи равна длине кинетической цепи: Р п = v.
При обрыве рекомбинацией (&) одна макромолекула образуется из двух кинетических цепей, и Р п = 2v. При смешанном обрыве (& ор + к ол) длина материальной цепи также не совпадает с длиной кинетической цепи:
Выведем уравнение для степени полимеризации из кинетических данных. Если полимеризация протекает в условиях квазистационарности при отсутствии ингибитора, то при достаточно малой глубине превращения, когда полимера в системе еще мало и, следовательно, скоростью передачи цепи на полимер и расходом мономера можно пренебречь:
где V a - скорость бимолекулярного обрыва цепи; - сумма скоростей передачи цени на мономер М и растворитель S;
При рекомбинации двух радикалов образуется одна материальная цепь, т.е. происходит среднестатистическое удваивание Р п, поэтому в знаменателе уравнения (5.13) перед членом, соответствующим обрыву путем рекомбинации, необходимо учесть сомножитель 0,5. Если обозначить долю полимерных радикалов, обрывающихся по механизму диспропорционирования, X, то доля радикалов, гибнущих при рекомбинации, равна (1 - X) и уравнение для Р п примет вид
Тогда для величины, обратной Р п,
получим

Выразив концентрацию радикала через скорость полимеризации и используя величины С м и C s , окончательно получаем
Полученное уравнение связывает среднечисловую степень полимеризации со скоростью реакции, константами передачи и концентрациями мономера и передающего агента. Из уравнения (5.15) следует, что степень полимеризации прямо пропорциональна концентрации мономера, обратно пропорциональна концентрации инициатора в степени 1/2, а максимальная степень полимеризации образующегося полимера при отсутствии других передающих агентов определяется реакцией передачи цепи на мономер (С м).
k" /2 k / k обр 2 = k p [M] / k обр = k n / = k n I / [I] 0,5 Иными словами, степень полимеризации и, следовательно, средняя молекулярная масса полимера при свободнорадикальной полимеризации обратно пропорциональна квадратному корню из концентрации инициатора.
Влияние различных факторов на процесс радикальной полимеризации.
1. Влияние температуры С повышением температуры увеличивается скорость реакции образования активных центров и реакции роста цепи. Таким образом, повышается суммарная скорость образования полимера. Обычно скорость полимеризации возрастает в 2-3 раза при повышении температуры на 10 ˚С. Однако при общем увеличении концентрации радикалов увеличивается и вероятность их столкновения друг с другом (обрыв цепи путем диспропорционирования или рекомбинации) или с низкомолекулярными примесями. В результате молекулярная масса полимера в целом уменьшается (средняя степень полимеризации уменьшается с ростом температуры), увеличивается доля низкомолекулярных фракций в полимере. Возрастает число побочных реаций, приводящих к образованию разветвленных молекул. Увеличивается нерегулярность при построении цепи полимера вследствие возрастания доли типов соединения мономера «голова к голове» и «хвост к хвосту». 2. Влияние концентрации инициатора.С повышением концентрации инициатора число свободных радикалов увеличивается, возрастает число активных центров, увеличивается суммарная скорость полимеризации.
Однако при общем увеличении концентрации радикалов увеличивается и вероятность их столкновения друг с другом, т.е. обрыва цепи, что приводит к снижению молекулярной массы полимера. 3. Влияние концентрации мономера.
При полимеризации в среде растворителя суммарная скорость полимеризации и молекулярная масса образующегося полимера увеличивается с повышением концентрации мономера. При полимеризации в инертном растворителе, не участвующем в реакции, скорость полимеризации равна  (часто x = 1,5). Большинство растворителей участвуют в полимеризации (в реакции передачи цепи). Поэтому получаются гораздо более сложные зависимости. 4. Влияние давления.
Давление высокое и сверхвысокое 300-500 МПа (3000-5000 ат) и выше значительно ускоряет полимеризацию. Пример.
Полимеризация метилметакрилата в присутствии
(часто x = 1,5). Большинство растворителей участвуют в полимеризации (в реакции передачи цепи). Поэтому получаются гораздо более сложные зависимости. 4. Влияние давления.
Давление высокое и сверхвысокое 300-500 МПа (3000-5000 ат) и выше значительно ускоряет полимеризацию. Пример.
Полимеризация метилметакрилата в присутствии  воздуха при 100˚C и p = 0,1 МПа продолжается 6 часов, под р = 300 МПа – 1 час, т.е. суммарная скорость полимеризации возрастает примерно в 6 раз. Аналогичным образом влияние p
сказывается на полимеризации стирола, винилацетата, изопрена и др. NB
!
Особенностью полимеризации под p
является то, что увеличение скорости не сопровождается уменьшением молекулярной массы получаемого полимера.
воздуха при 100˚C и p = 0,1 МПа продолжается 6 часов, под р = 300 МПа – 1 час, т.е. суммарная скорость полимеризации возрастает примерно в 6 раз. Аналогичным образом влияние p
сказывается на полимеризации стирола, винилацетата, изопрена и др. NB
!
Особенностью полимеризации под p
является то, что увеличение скорости не сопровождается уменьшением молекулярной массы получаемого полимера.
Ингибиторы и регуляторы полимеризации
Явления обрыва и передачи цепи широко используются на практике для:
- предотвращения преждевременной полимеризации при хранении мономеров;
- для регулирования процесса полимеризации
 ис.2 Термическая полимеризация стирола при 100 ˚С в присутствии ингибиторов и замедлителей: 1 – без добавок; 2- 0,1% бензохинона (ингибитор); 3 – 0,2% нитробензола (ингибитор); 4 – 0,5% нитробензола (замедлитель)
ис.2 Термическая полимеризация стирола при 100 ˚С в присутствии ингибиторов и замедлителей: 1 – без добавок; 2- 0,1% бензохинона (ингибитор); 3 – 0,2% нитробензола (ингибитор); 4 – 0,5% нитробензола (замедлитель)Для регулирования процесса полимеризации применяют ингибиторы и замедлители полимеризации. Ингибиторы
– низкомолекулярные вещества, которые меняют длительность индукционного периода, замедляя его. Это часто необходимо делать в технологии производства полимеров для предотвращения преждевременной полимеризации в неконтролируемых условиях. Ингибиторы: хиноны, ароматические амины, нитросоединения, фенолы, органические соли  ,
,  ,
,  ,
,  и т.д. Пример
: гидрохинон Хинон взаимодействует со свободными радикалами, превращая их в неактивные продукты. Гибель радикалов увеличивает длину индукционного периода. Наряду с ингибиторами, позволяющими полностью остановить полимеризацию, существуют замедлители полимеризации
, которые только уменьшают её скорость. Замедлитель
выполняет двойную роль: снижает концентрацию радикалов и уменьшает время их жизни, что приводит к снижению длины полимерной цепи. Ингибитор не влияет на скорость полимеризации, но предотвращает начало инициирования цепи, увеличивая индукционный период на кинетической кривой полимеризации. Длительность индукционного периода обычно пропорциональна количеству введенного ингибитора. Одно и то же вещество может выступать и как ингибитор, и как замедлитель, и как регулятор полимеризации в зависимости от природы полимеризуемого мономера. Например, кислород, который замедляет полимеризацию винилацетата и ускоряет полимеризацию стирола. При больших давлениях и высоких температурах кислород способствует полимеризации этилена. Это явление используют при промышленном производстве полиэтилена высокого давления. Кислород образует пероксиды или гидропероксиды при взаимодействии с мономерами или растущими цепями. гидропероксид пероксид В зависимости от стабильности промежуточных пероксидов или гидропероксидов они могут либо увеличивать концентрацию радикалов и ускорять полимеризацию, либо дезактивировать имеющиеся радикалы и замедлять или даже ингибировать полимеризацию. Рис.1.3 с.28 кулезнев Пример: ароматические нитро- и нитрозосоединения. Регуляторы полимеризации
вызывают преждевременный обрыв материальной цепи
, снижая молекулярную массу полимера пропорционально введенному количеству регулятора. Примером их являются меркаптаны, в том числе додецилмеркаптан. Из-за большой длины углеводородной цепи его молекулы недостаточно активны и расходуются медленно.
и т.д. Пример
: гидрохинон Хинон взаимодействует со свободными радикалами, превращая их в неактивные продукты. Гибель радикалов увеличивает длину индукционного периода. Наряду с ингибиторами, позволяющими полностью остановить полимеризацию, существуют замедлители полимеризации
, которые только уменьшают её скорость. Замедлитель
выполняет двойную роль: снижает концентрацию радикалов и уменьшает время их жизни, что приводит к снижению длины полимерной цепи. Ингибитор не влияет на скорость полимеризации, но предотвращает начало инициирования цепи, увеличивая индукционный период на кинетической кривой полимеризации. Длительность индукционного периода обычно пропорциональна количеству введенного ингибитора. Одно и то же вещество может выступать и как ингибитор, и как замедлитель, и как регулятор полимеризации в зависимости от природы полимеризуемого мономера. Например, кислород, который замедляет полимеризацию винилацетата и ускоряет полимеризацию стирола. При больших давлениях и высоких температурах кислород способствует полимеризации этилена. Это явление используют при промышленном производстве полиэтилена высокого давления. Кислород образует пероксиды или гидропероксиды при взаимодействии с мономерами или растущими цепями. гидропероксид пероксид В зависимости от стабильности промежуточных пероксидов или гидропероксидов они могут либо увеличивать концентрацию радикалов и ускорять полимеризацию, либо дезактивировать имеющиеся радикалы и замедлять или даже ингибировать полимеризацию. Рис.1.3 с.28 кулезнев Пример: ароматические нитро- и нитрозосоединения. Регуляторы полимеризации
вызывают преждевременный обрыв материальной цепи
, снижая молекулярную массу полимера пропорционально введенному количеству регулятора. Примером их являются меркаптаны, в том числе додецилмеркаптан. Из-за большой длины углеводородной цепи его молекулы недостаточно активны и расходуются медленно.

 ,
,  .
. Способы проведения полимеризации
Радикальную полимеризацию проводят в блоке (массе), растворе, эмульсии, суспензии и газовой фазе. При этом процесс может протекать в гомогенных или гетерогенных условиях. Кроме того, фазовое состояние исходной реакционной смеси может также меняться в ходе полимеризации.
Полимеризация в блоке (в массе )
Полимеризация в растворе
В отличие от полимеризации в блоке в данном случае отсутствуют местные перегревы, так как тепло реакции снимается растворителем, выполняющим также роль разбавителя. Уменьшается вязкость реакционной системы, что облегчает её перемешивание.
Однако возрастает роль (доля) реакций передачи цепи, что приводит к понижению молекулярной массы полимера. Кроме того, полимер может быть загрязнён остатками растворителя, который не всегда удаётся удалить из полимера. Существует два способа проведения полимеризации в растворе. а) Применяют растворитель, в котором растворяется и мономер, и полимер. Получаемый полимер используют непосредственно в растворе или выделяют его осаждением или испарением растворителя. б) В растворителе, используемом для полимеризации, растворяется мономер, но не растворяется полимер. Полимер по мере образования выпадает в твердом виде и может быть отделен фильтрованием.
Полимеризация в суспензии (бисерная или гранульная)
 в виде мелких капель. Устойчивость дисперсии достигается механическим перемешиванием и введением в реакционную систему специальных добавок – стабилизаторов. Процесс полимеризации осуществляют в каплях мономера, которые можно рассматривать как микрореакторы блочной полимеризации. Применяют инициаторы, растворимые в мономере. Достоинством этого процесса является хороший отвод тепла, недостатком - возможность загрязнения полимера остатками стабилизатора
в виде мелких капель. Устойчивость дисперсии достигается механическим перемешиванием и введением в реакционную систему специальных добавок – стабилизаторов. Процесс полимеризации осуществляют в каплях мономера, которые можно рассматривать как микрореакторы блочной полимеризации. Применяют инициаторы, растворимые в мономере. Достоинством этого процесса является хороший отвод тепла, недостатком - возможность загрязнения полимера остатками стабилизатора Полимеризация в эмульсии (эмульсионная полимеризация)
Газофазная полимеризация
- Свободнорадикальная полимеризация – один из видов цепных процессов синтеза полимеров. Поляризация исходных молекул мономера облегчает их реакции с радикалами инициатора при химическом инициировании или при физических методах генерации радикалов. Электроноакцепторные заместители способствуют большей стабильности радикалов мономера и растущих цепей. Процесс радикальной полимеризации можно регулировать различными приемами как по скорости конверсии мономера, так и по величине молекулярной массы полимера. Для этого используют добавки низкомолекулярных веществ, выполняющих функции ингибиторов или замедлителей реакции, а также осуществляющих передачу реакционной цепи или снижающих энергию активации распада инициаторов на радикалы. Знание закономерностей свободнорадикальной полимеризации позволяет управлять структурой полимера, а следовательно, и его физическими и механическими свойствами. Благодаря простоте этот способ получения полимеров нашел широкое применение в промышленности.

